
You can read the press release here, or our piece in the Conversation, We’ve unveiled the waratah’s genetic secrets, helping preserve this Australian icon for the future.
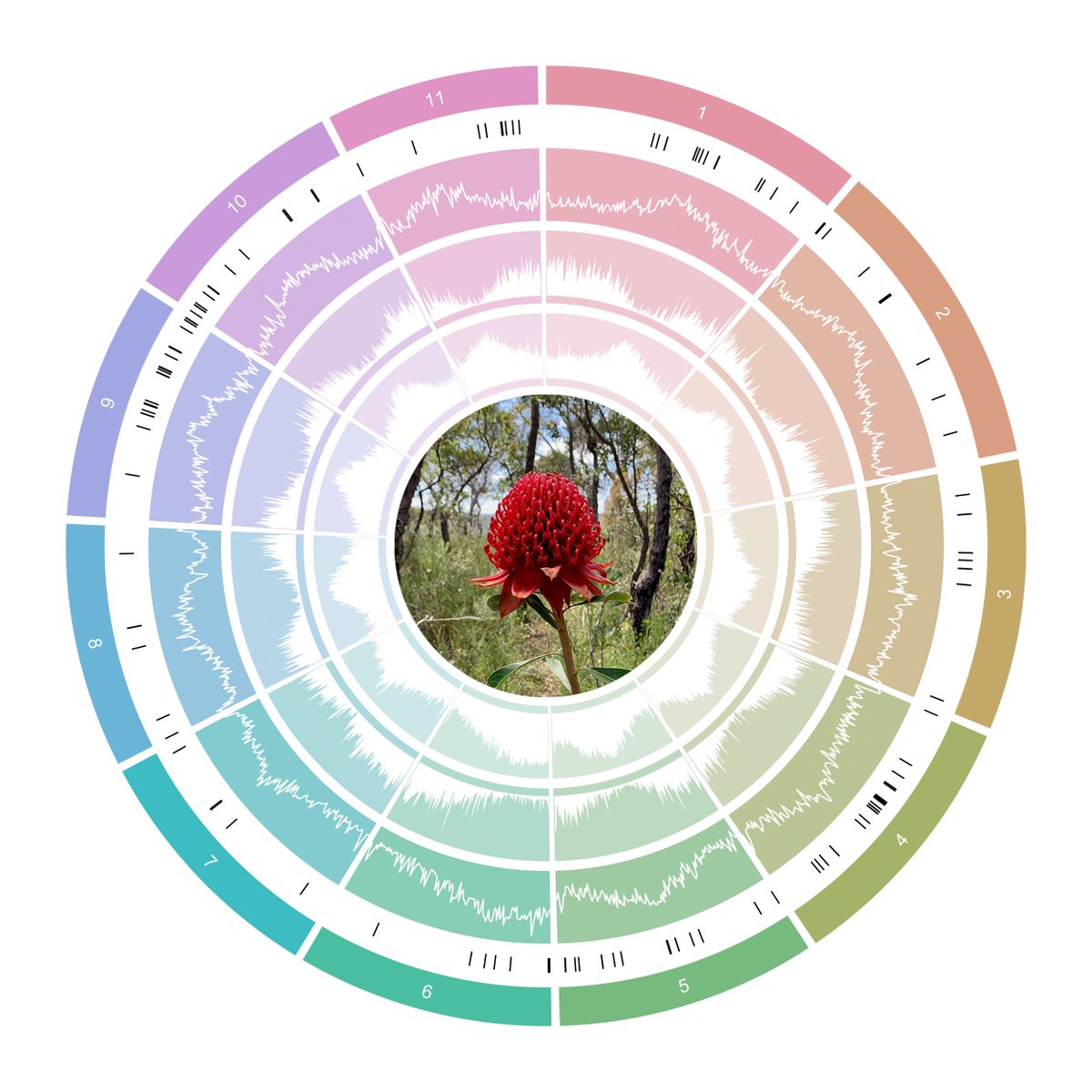
In this paper, we present a chromosome-level assembly for the NSW State Floral Emblem, the New South Wales waratah, Telopea speciosissima. This joins macadamia as the 2nd reference genome for the Proteaceae family & should help future studies for the remaining ca. 1700 species.
The genome was assembled from a ONT chassis, scaffolded with 10x Genomics linked reads and Phase Genomics HiC - made possible thanks to quality data from AGRF and the Ramaciotti Centre for Genomics. The final assembly was chromosome-level, with 94.1% on the 11 chromosomes (2n = 22).
As well as the assembly itself, the paper presents a three genomics tools that we hope will be helpful for other assemblies:
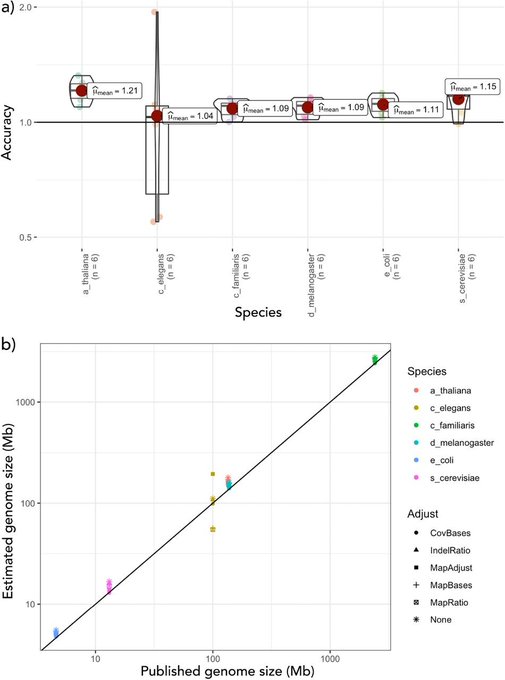
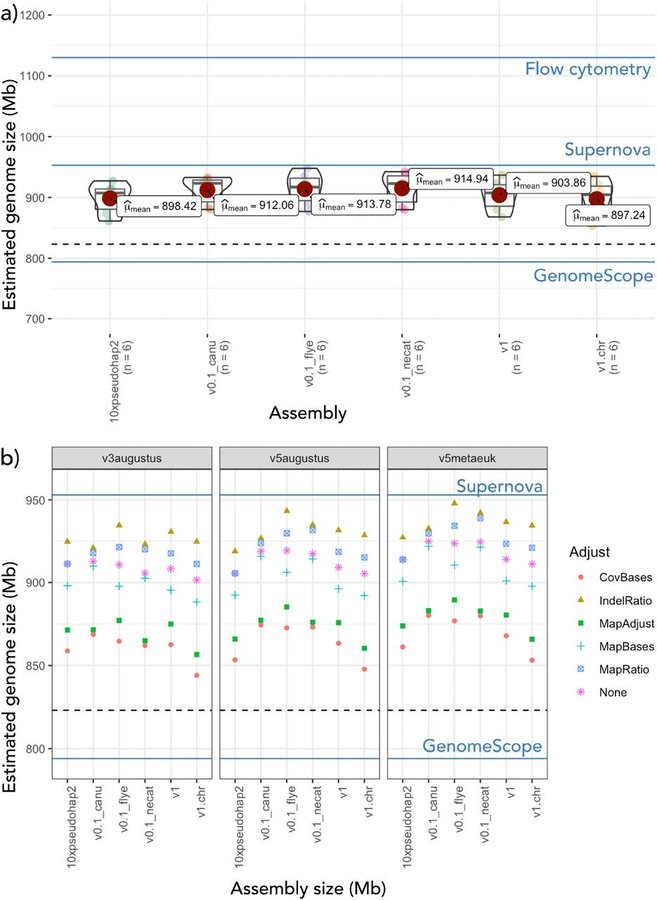
1. DepthSizer uses long-read depths and BUSCO predictions to estimate genome size. We estimated the waratah genome to be ca. 900 Mbp - bigger than kmer estimates, but smaller than flow cytometry of Tasmanian waratah.
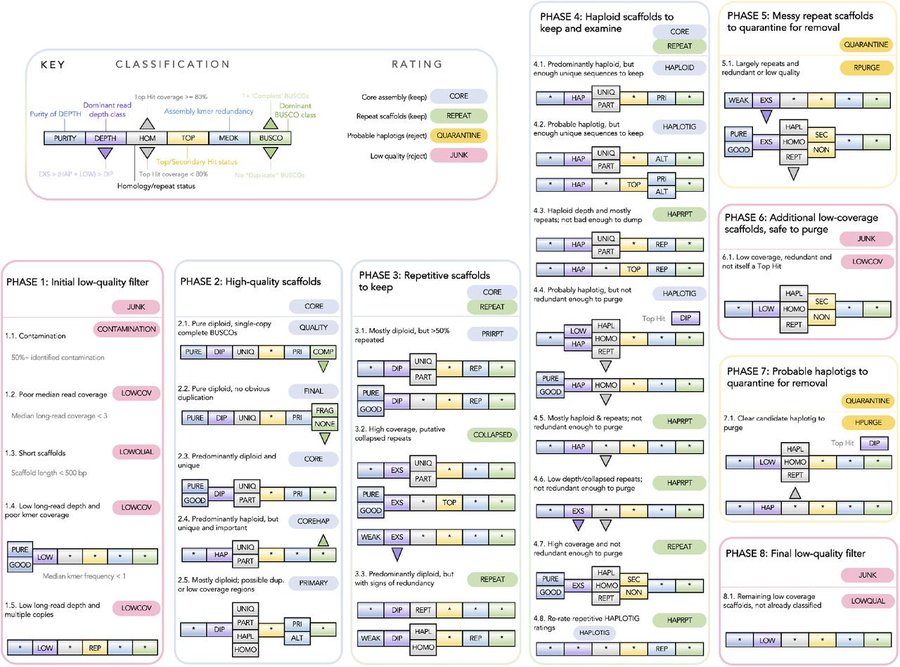
2. Diploidocus builds on Purge Haplotigs, combining read depths, kmer frequencies & BUSCO predictions to classify and curate/filter assembly scaffolds. This decreases false duplications & contamination, and flags collapsed repeats for closer inspection.
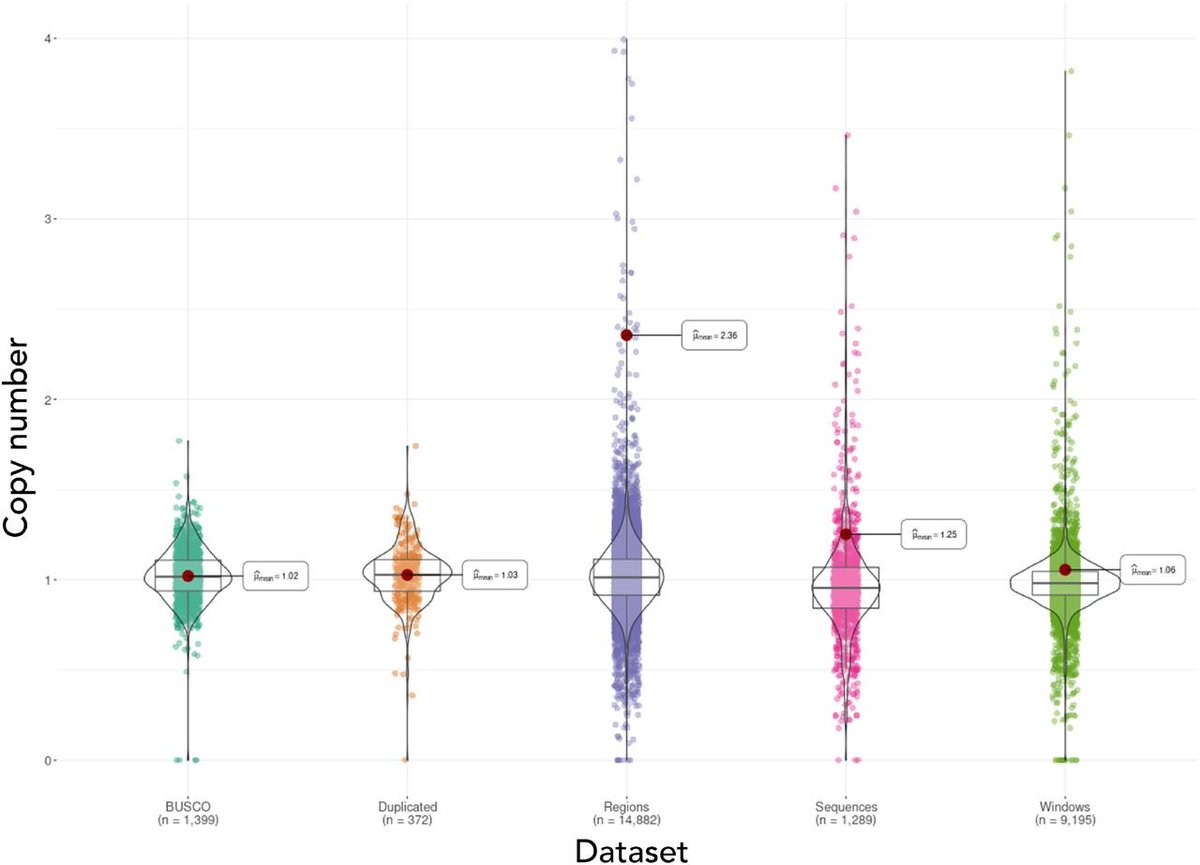
3. DepthKopy uses BUSCO Complete genes to establish sequencing depth (like DepthSizer) and then estimates copy number for regions (e.g. genes), scaffolds & sliding windows of the assembly. This showed that most “Duplicated” BUSCOs are real duplicates.
Chen SH, Rossetto M, van der Merwe M, Lu-Irving P, Yap JS, Sauquet H, Bourke G, Amos TG, Bragg JG & Edwards RJ (accepted): Chromosome-level de novo genome assembly of Telopea speciosissima (New South Wales waratah) using long-reads, linked-reads and Hi-C. Molecular Ecology Resources.
[Mol Ecol Res]
[bioRxiv]
Abstract
Telopea speciosissima, the New South Wales waratah, is an Australian endemic woody shrub in the family Proteaceae. Waratahs have great potential as a model clade to better understand processes of speciation, introgression and adaptation, and are significant from a horticultural perspective. Here, we report the first chromosome-level genome for T. speciosissima. Combining Oxford Nanopore long-reads, 10x Genomics Chromium linked-reads and Hi-C data, the assembly spans 823 Mb (scaffold N50 of 69.0 Mb) with 97.8% of Embryophyta BUSCOs “Complete”. We present a new method in Diploidocus (https://github.com/slimsuite/diploidocus) for classifying, curating and QC-filtering scaffolds, which combines read depths, k-mer frequencies and BUSCO predictions. We also present a new tool, DepthSizer (https://github.com/slimsuite/depthsizer), for genome size estimation from the read depth of single-copy orthologues and estimate the genome size to be approximately 900 Mb. The largest 11 scaffolds contained 94.1% of the assembly, conforming to the expected number of chromosomes (2n = 22). Genome annotation predicted 40,158 protein-coding genes, 351 rRNAs and 728 tRNAs. We investigated CYCLOIDEA (CYC) genes, which have a role in determination of floral symmetry, and confirm the presence of two copies in the genome. Read depth analysis of 180 “Duplicated” BUSCO genes using a new tool, DepthKopy (https://github.com/slimsuite/depthkopy), suggests almost all are real duplications, increasing confidence in the annotation and highlighting a possible need to revise the BUSCO set for this lineage. The chromosome-level T. speciosissima reference genome (Tspe_v1) provides an important new genomic resource of Proteaceae to support the conservation of flora in Australia and further afield.
If you want a read and don’t have access, please get it touch or check out the bioRxiv preprint.
No comments:
Post a Comment