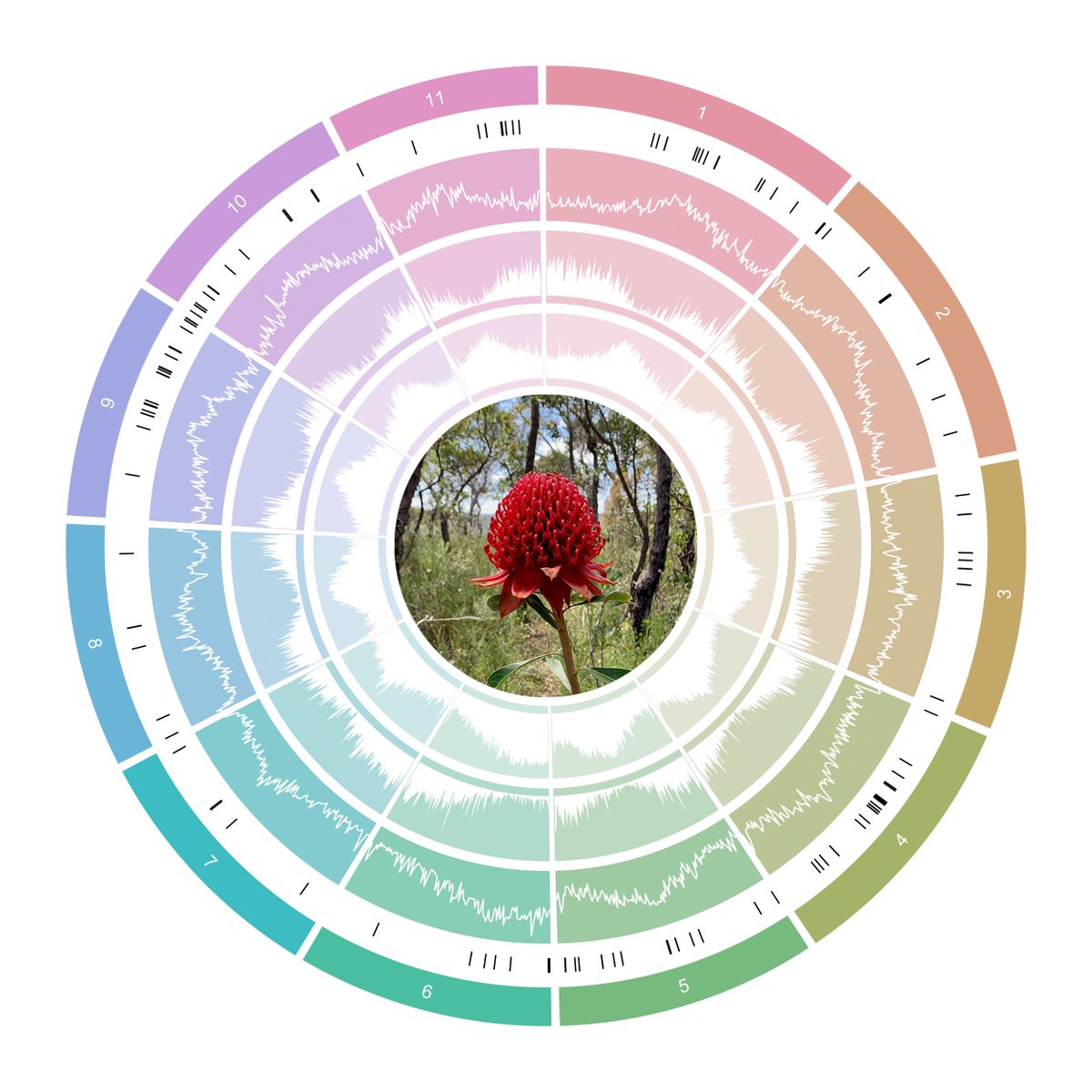
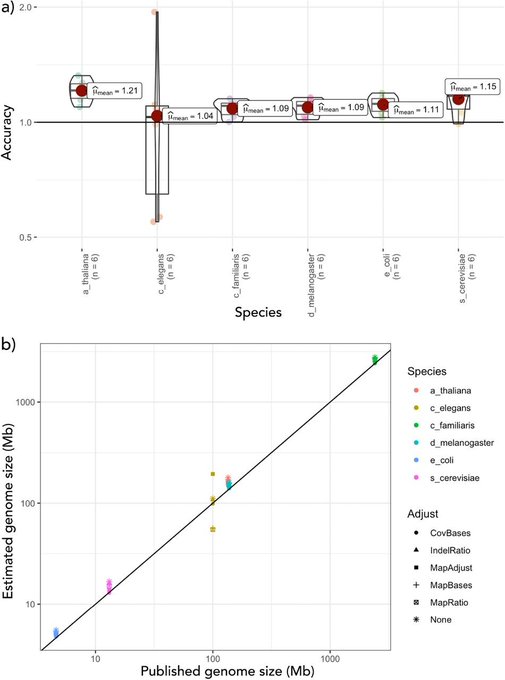
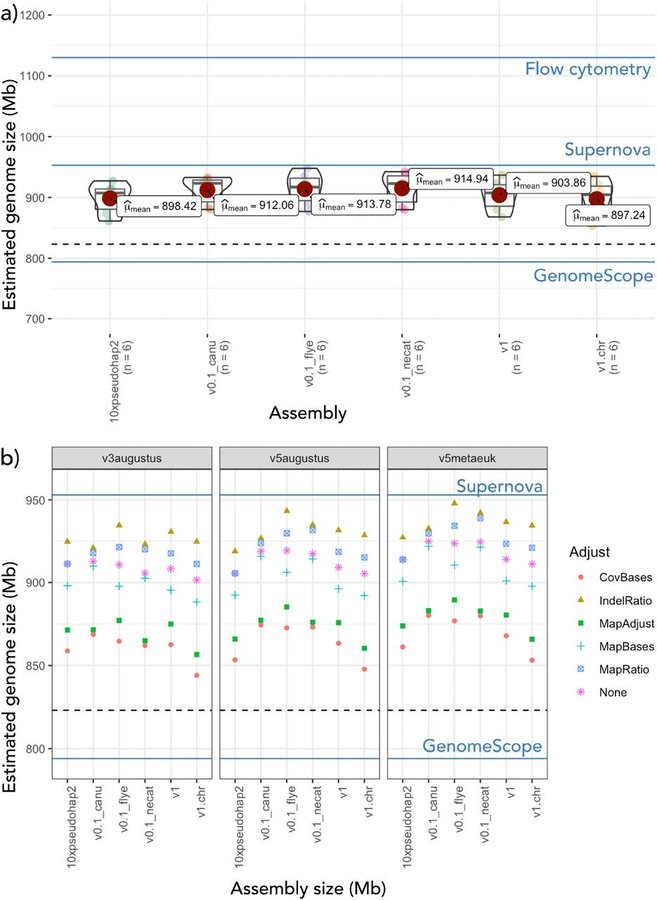
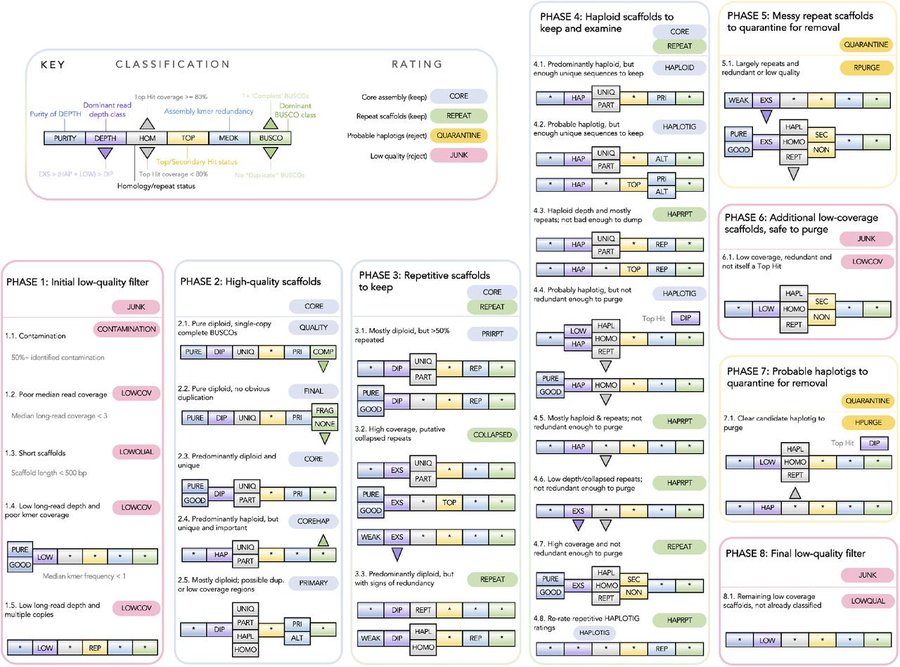
The first of the BABS Genome papers has finally arrived, featuring our two 10x Genomics Supernova snake genomes. Such is the speed that genomics is moving, the snake assemblies themselves have moved on quite a bit since then and we hope to release chromosome-level versions soon. (The goalposts for a genome paper moved faster than they could be written up - always a challenge without dedicated researchers working on assemblies! Do get in touch if they’d be useful and we can collaborate.)
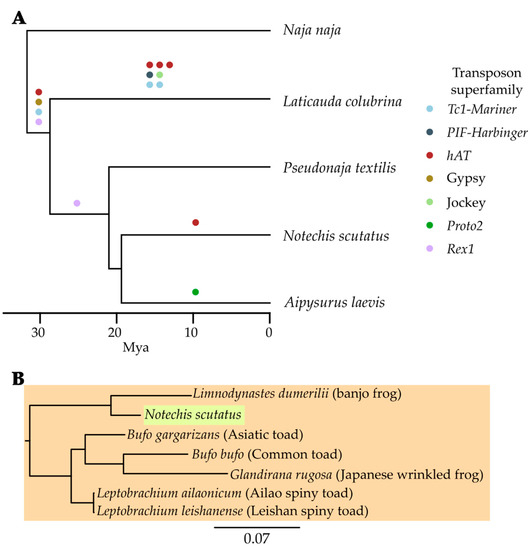 Rather than a pure genome paper, this paper makes use of our two elapid genomes to ask some interesting questions about possible horizontal transfer of transposable (mobile genetic) elements during the evolution of sea snakes - our two elapids provided good sister (mainland tiger snake) and outgroup (eastern brown snake) taxa for the olive sea snake, which was the focus of the study. It was doubly pleasing to collaborate on a transposable elements paper, as they were the subject of my PhD (albeit in bacteria, see here and here).
Rather than a pure genome paper, this paper makes use of our two elapid genomes to ask some interesting questions about possible horizontal transfer of transposable (mobile genetic) elements during the evolution of sea snakes - our two elapids provided good sister (mainland tiger snake) and outgroup (eastern brown snake) taxa for the olive sea snake, which was the focus of the study. It was doubly pleasing to collaborate on a transposable elements paper, as they were the subject of my PhD (albeit in bacteria, see here and here).
This paper is part of a special issue, Mobile Elements in Phylogenomic Reconstructions, and features some interesting examples of probable horiztonal transfer of mobile elements that provide insights into the evolutionary history of these species.
Galbraith JD, Ludington AJ, Sanders KL, Amos TG, Thomson VA, Enosi Tuipulotu D, Dunstan N, Edwards RJ, Suh A, Adelson DL (2022): Horizontal transposon transfer and its implications for the ancestral ecology of hydrophiine snakes. Genes 13(2):217. [Genes] [PDF] [bioRxiv]
Abstract
Transposable elements (TEs), also known as jumping genes, are sequences able to move or copy themselves within a genome. As TEs move throughout genomes they often act as a source of genetic novelty, hence understanding TE evolution within lineages may help in understanding environmental adaptation. Studies into the TE content of lineages of mammals such as bats have uncovered horizontal transposon transfer (HTT) into these lineages, with squamates often also containing the same TEs. Despite the repeated finding of HTT into squamates, little comparative research has examined the evolution of TEs within squamates. Here we examine a diverse family of Australo–Melanesian snakes (Hydrophiinae) to examine if the previously identified, order-wide pattern of variable TE content and activity holds true on a smaller scale. Hydrophiinae diverged from Asian elapids ~30 Mya and have since rapidly diversified into six amphibious, ~60 marine and ~100 terrestrial species that fill a broad range of ecological niches. We find TE diversity and expansion differs between hydrophiines and their Asian relatives and identify multiple HTTs into Hydrophiinae, including three likely transferred into the ancestral hydrophiine from fish. These HTT events provide the first tangible evidence that Hydrophiinae reached Australia from Asia via a marine route.