Lorne Genome 2022 (the 43rd Annual Lorne Genome Conference 2022) kicks off today in Lorne and online. I wasn’t able to make it in person this year due to Omicron and teaching commitments, but happily the lab is still well represented. As well as an online talk, we have two in-person posters, so please check these out if you are lucky enough to be attending in the flesh.
Details below.
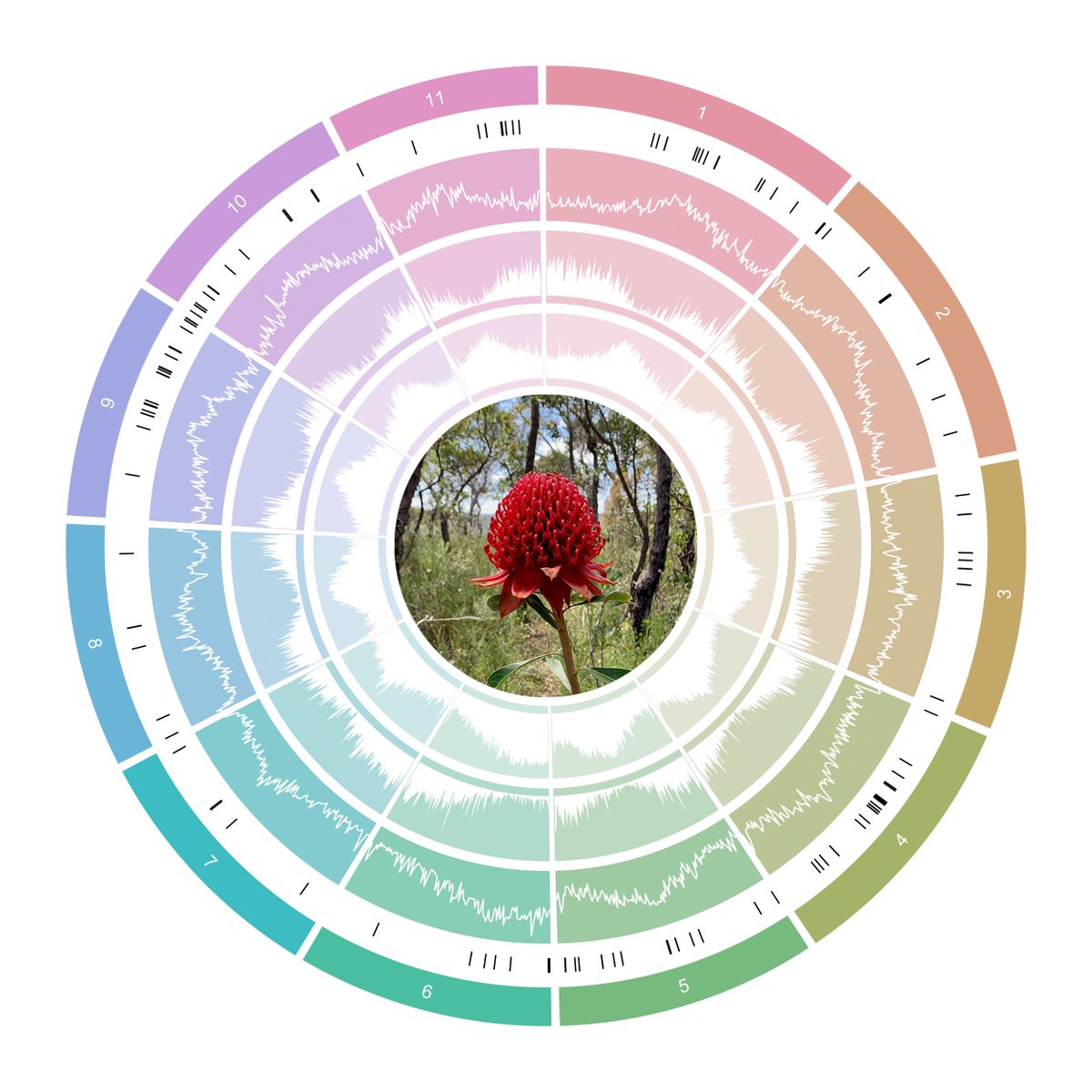
A chromosome-level reference genome for Telopea speciosissima (New South Wales waratah) provides insight into waratah evolution (#138)
Stephanie H Chen, Jason G Bragg, Richard J Edwards
Telopea is an eastern Australian genus of five species of long-lived shrubs in the family Proteaceae. Previous work has characterised population structure and patterns of introgression between Telopea species. These studies were performed using a limited set of genetic markers, but point to the great potential of waratah as a model clade for understanding the processes of divergence, environmental adaptation and speciation, when enhanced by a genome-wide perspective enabled by a reference genome. However, few Proteaceae genomes and no waratah genomes are available.
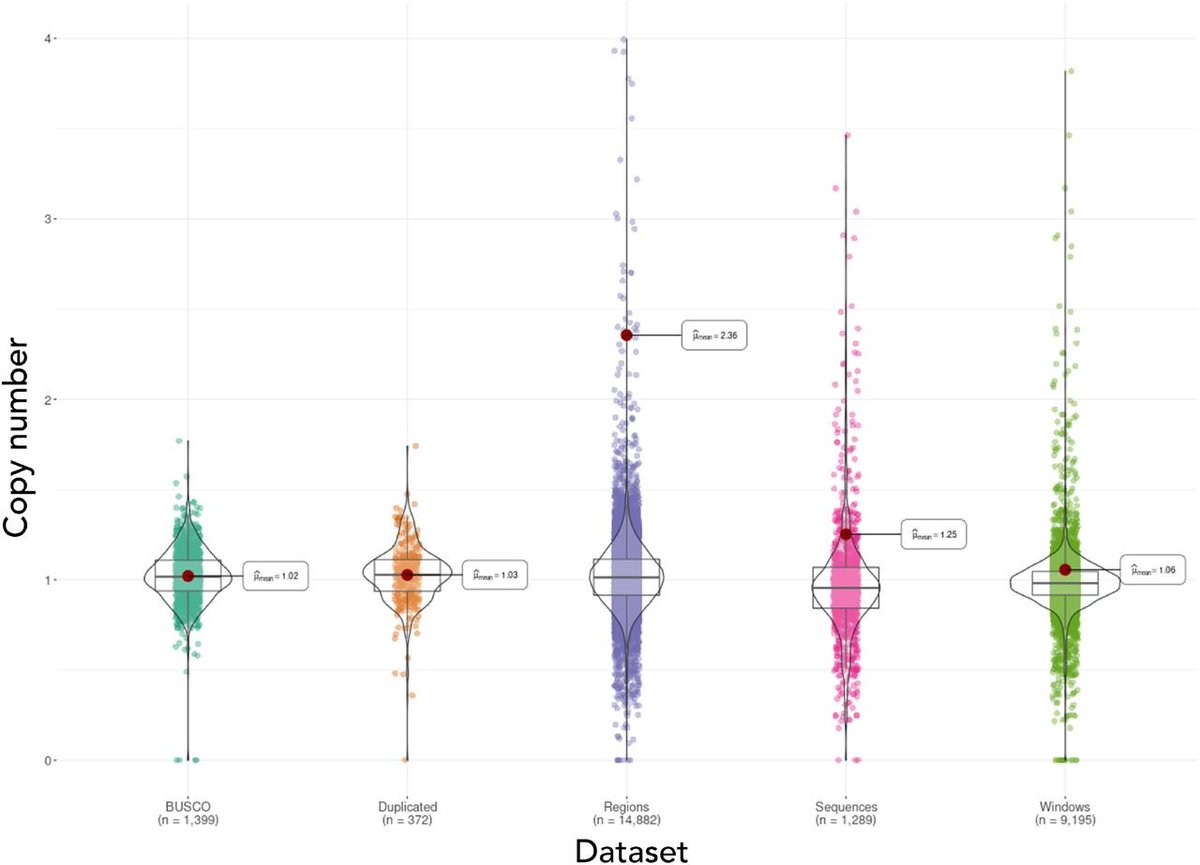
We assembled the first chromosome-level reference genome for T. speciosissima (New South Wales waratah; 2n = 22) using Nanopore long-reads, 10x Chromium linked-reads and Hi-C data. The assembly spans 823 Mb (scaffold N50 of 69.0 Mb) with 97.8 % of Embryophyta universal single-copy orthologues (BUSCOs; n = 1,614) complete. Read depth analysis of 140 ‘Duplicated’ BUSCO genes reveals that almost all are real duplications, increasing confidence in protein family analysis using annotated protein-coding genes, highlighting a possible need to revise the BUSCO set for this lineage. Genome annotation predicted 34,706 genes and pseudogenes, including 27,481 protein-coding genes. We examined the evolutionary dynamics of Telopea using the reference genome in conjunction with DArTseq (n = 244) and whole genome shotgun sequencing (n = 14) of each of the seven lineages; there are three lineages of T. speciosissima – coastal, upland and southern.
Here, I will discuss the population structure and demographic history of the genus. We also examined phylogenomic relationships and developed a scalable method of rapidly generating species trees from short-read data to maximise the recovery of informative data from genomic datasets. The waratah reference genome represents an important new genomic resource in Proteaceae to accelerate our understanding of the origins and evolutionary dynamics of the Australian flora.
[Read more about the waratah genome, here.]
Small but mitey: high-quality long-read assembly of a streamlined mite genome from contaminated sequencing data (#17)
Richard J Edwards, Stephanie H Chen, Jason G Bragg.

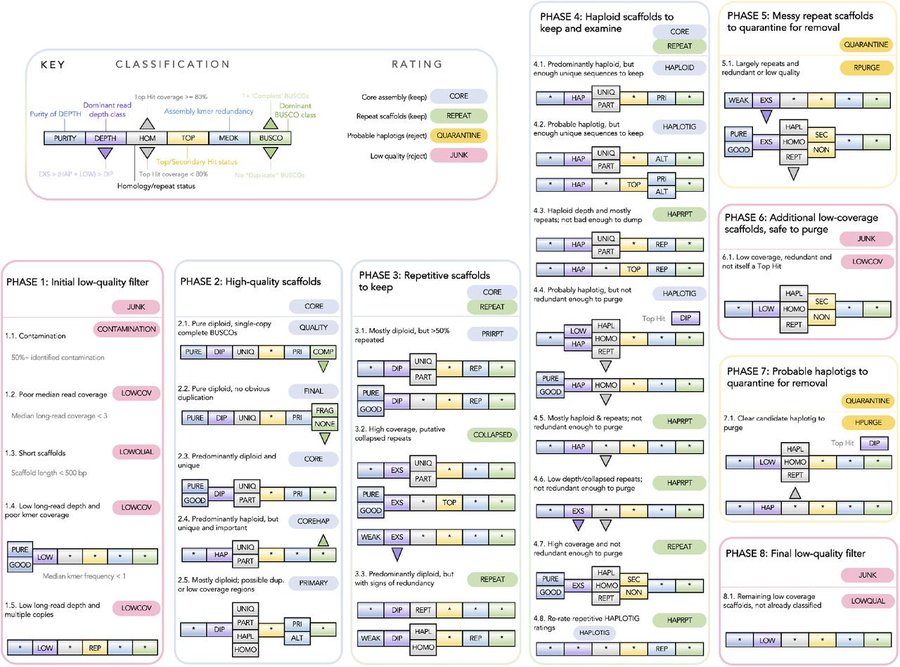
As pilot data for project on myrtle rust resistance, we previously assembled two Myrtaceae genomes using 10x Chromium linked reads: Rhodamnia argentea (silver malletwood) and Syzygium oleosum (blue lilly pilly). Both draft genomes achieved scaffolding (N50 > 850 kb) and completeness (BUSCOv3 embryophyta_odb9 > 90 %) of sufficient quality to be annotated by NCBI RefSeq. However, signs of arthropod sequence contamination were subsequently found in the Rhodamnia argentea assembly. We therefore sought to identify and eliminate this contamination during improvement and curation of the genome for publication.
A risk-averse analysis highlighted 49.6 Mb (11.95%) on 2,996 of 15,781 scaffolds of possible arthropod origin. An improved assembly of the same tree, incorporating ~50X long-read (ONT) sequencing, has confirmed this contamination as 11 scaffolds (34.6 Mb) that are distinct from 75 R. argentea assembly scaffolds (346.7 Mb), increasing the likelihood of contamination over the integration of horizontally transferred genes. Taxonomic analysis of predicted protein-coding genes using Taxolotl (https://github.com/slimsuite/taxolotl) suggested that the contamination most likely originates from some form of mite (Order: Trombidiformes), but limited NCBInr mite sequences precluded better taxonomic resolution. Curiously, these contamination scaffolds showed a high depth of coverage (~36X), but a fairly low BUSCO completeness of 58.1% (v5 Augustus, metazoa_odb10 n=954), apparently inconsistent with typical mite genomes.
Phylogenomic analysis with available mite genomes identified the closest relative as Aculops lycopersici, a microscopic (0.2 mm long) eriophyoid mite with a heavily streamlined 32.5 Mb genome. Original low completeness appears to be from a combination of genome reduction and poor performance of that BUSCO version; BUSCO v5 MetaEuk eukaryota_odb10 (n=255) reports 82.8% completeness, which is approaching the 86.3% of A. lycopersici. Here, we discuss the evidence that we have assembled a highly complete but streamlined genome from an unknown eriophyoid mite, plus the need to improve genomic representation of contaminating pest species.
A genetic perspective on rapid adaptation in the globally invasive European starling (Sturnus vulgaris) (#255)
Katarina C Stuart, Richard J Edwards, William (Bill) B Sherwin, Lee Ann Rollins.
Few invasive birds are as globally successful or as well-studied as the common starling (Sturnus vulgaris). Native to the Palaearctic, the starling has been a prolific invader in North and South America, southern Africa, Australia, and The Pacific Islands, while facing declines in excess of 50% in in some native regions. Starlings present an invaluable opportunity to test predictions about the evolutionary trajectory of invasive populations, and gain insight into genetic shifts in response to anthropogenic alteration and climate change.
My research focuses primarily on the invasive European starling population in Australia and aims to investigate the genetics underlying their evolution, using a range of genomic approaches. Through historic museum sample sequencing, I examine single nucleotide polymorphism variations shifts between the native range and Australia, and find parallel selection on both continents, possibly resulting from common global selective forces such as exposure to pollutants and carbohydrate exposure. I further examine matched genetic, morphological, and environmental data to reveal patterns of heritability and plasticity across ecologically significant phenotypic traits, revealing that elevation, as well as rainfall and temperature variability plays an important role in shaping morphology and genetics. Finally, I investigated patterns of structural variants, to uncover evolutionarily significant large-scale genetic variants across a global data set, and more specifically characterise their role in rapid starling adaptation across the entirety of the Australian range. Overall, my research seeks to better understand mechanisms and patterns of genetic change within this species, which may be used to inform invasion or native range management. More broadly, this evolutionary research into the starling provide an important perspective on the role of rapid evolution in invasive species persistence, and the global pressures that may shape range shifts and evolution across many similar avian taxa.