Yesterday’s excellent Australian Pathogen Bioinformatics Symposium 2014 (#APBS2014) reminded me that this post was a bit overdue: originally posted at the UNSW Science Wavelength blog on October 30, 2014. Following APBS2014, we are also looking into how we can identify which other pathogens might be using a similar strategy.
Every living cell is packed full of tiny molecular machines. Many of these machines are made from proteins: assembled chains of amino acid building blocks, which twist, wrap and fold up into complex three-dimensional shapes. The way that these machines interact with each other, and other molecules in the cell, ultimately determines how a cell behaves, divides (or not) and dies.
Viruses hijack this cellular machinery and reprogram it to their own ends using their own set of proteins. Many viruses are tiny, with genomes that encode only a handful of proteins, and yet they need to take control of a multitude of host processes. The way that they achieve this is by mimicking small host interaction motifs called SLiMs (short linear motifs), which are the focus of study in my lab.
For example, some viruses use the host DNA replication machinery for their own replication and mimic a SLiM that interacts with an important cell cycle checkpoint protein called retinoblastoma. By mimicking the retinoblastoma-binding motif, the virus tricks the cell into entering the DNA replication phase of the cell cycle. Other SLiMs are mimicked to prevent host cells committing suicide through a process called apoptosis, or to hijack cellular transport machinery and get the viral proteins to the right place.
My lab is a bioinformatics group, using computational tools to analyse biological data. As their name suggests, SLiMs are very short regions of the protein chain that have a small number of specific amino acids in specific linear configuration that enables them to interact with particular proteins in the cell. Not all protein folds up into complex shapes; SLiMs are generally found in regions that remain in a flexible, linear “intrinsically disordered” state. This means that we can potentially identify SLiMs by careful examination of the protein “sequence”: the precise composition and order of amino acids from a pool of twenty common types, as encoded by the DNA. This is challenging because one stretch of protein looks much like any other when written as a string of letters. In reality, these letters represent chemical groups with distinct properties and so our tools also consider evolutionary and structural signals in the data.
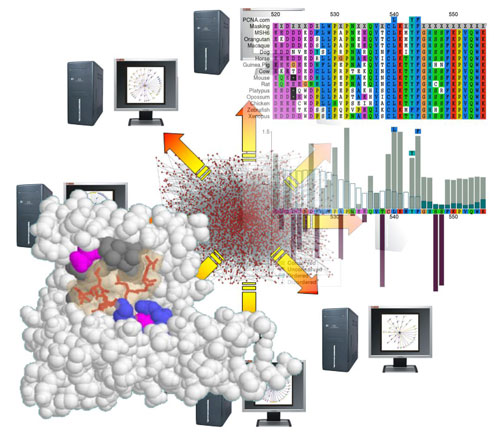 My background is in genetics and I consider myself an evolutionary biologist at heart. The famous adage of Theodosius Dobzhansky, “Nothing in biology makes sense except in the light of evolution” is particularly true of sequence analysis. The evolutionary process leaves clues in the sequences and it is those clues that my lab seeks to exploit.
My background is in genetics and I consider myself an evolutionary biologist at heart. The famous adage of Theodosius Dobzhansky, “Nothing in biology makes sense except in the light of evolution” is particularly true of sequence analysis. The evolutionary process leaves clues in the sequences and it is those clues that my lab seeks to exploit.
Many people are familiar with the concept of evolutionary conservation, in which functionally important traits (or sequences) are maintained through time by natural selection. Mutations of these sites are detrimental to reproductive success and do not have a long-term future in the population. By aligning and comparing proteins that share ancestry, it is possible to identify regions and sites under such constraint as those that have not changed. However, there is an important time component with such analyses; whilst some sites will be unchanged due to constraints, others may simply reflect shared ancestry with insufficient time to accumulate mutations. Conservation alone also says nothing about the function of the conserved site and there are many functional and structural constraints that would drown out any signal left by very small functional elements like SLiMs.
For SLiM discovery, we also harness a second evolutionary process: “convergent” evolution, in which the same trait independently arises on a different background. In the context of SLiMs, a specific configuration of amino acids is independently acquired by proteins that do not share identifiable common ancestry. Because SLiMs are so small and involve so few positions, a single amino acid substitution can create (or destroy) a SLiM in a protein, which is precisely how viruses are able to stumble across useful host motifs. If useful, such accidental discoveries are captured by natural selection.
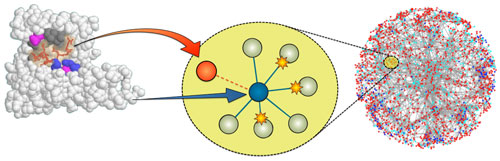
We identify candidates for convergent evolution as sequence patterns that are over-represented in a set of otherwise unrelated proteins that interact with a common partner protein. To identify molecular mimicry, we make use of experimental data in which a viral protein has been shown to interact with a human target protein. We look for sequence patterns that are found in the viral protein and enriched in the human proteins that interact with the target.
We still have a long way to go but if we can work out how viruses are disrupting the cellular machinery of their hosts, we might be able to work out how to stop them.









